2月6日,中国科学院昆明动物研究所吕雪梅、张亚平课题组与北京大学生命科学学院陆剑课题组、中国疾病预防控制中心谭文杰课题组合作在《科学通报》上发表了题为“Evolutionary analysis and lineage designation of SARS-CoV-2 genomes”的论文,解析了新型冠状病毒基因组的演化规律,提出了具有层次结构的谱系/亚谱系划分系统。
病毒的演化极其迅速,随着感染人数爆发性增加,新冠病毒会快速发展出许多不同的株系。探明病毒的谱系发生历史,对不同的毒株进行分型,结合流行病学及临床数据揭示新冠病毒的变异规律,对疫情的防控尤为重要。在研究团队前期的工作中,已根据两个高度连锁的位点将新冠病毒划分为L和S两个主要谱系,并描述了这两个谱系在传播过程中不同的行为模式。国际上也已发表诸多研究为新冠病毒进行分型。然而,随着新冠病毒基因组序列的迅速积累,人们又检测到并发表了成千上万的基因组变异,出现了许多值得进一步标记的亚型,大量相似的病毒序列对SARS-CoV-2基因组系统发育的可靠推断提出了重大挑战,急需对这些新出现的变异进行梳理与描述,更加精细化地展示新冠病毒在人群中的演变轨迹。
为解决这一问题,研究团队分析了121,618个高质量的病毒基因组中的单核苷酸变异(SNV),在L和S分型的基础上,根据3个主导性SNV将L谱系划分为L1和L2两个主要亚谱系,再根据位点的突变频率和连锁性筛选出201个代表性SNV,进一步逐级划分出130个亚谱系(S中37个,L1中35个,L2中58个),绘制成完整的反映各个谱系之间亲缘关系的单倍型网络图(图1)。作者同时建立了实时更新的配套网站(www.covid19evolution.net)方便查看亚谱系信息,此外用户还可自由上传新冠病毒基因组序列进行谱系鉴定。
为阐明不同亚谱系的病毒的流行程度是否有所不同,作者依据上述谱系划分系统对有具体采样时间信息的119,168个高质量病毒基因组进行划分,并挖掘了这些谱系的时空分布演变规律。研究发现,各个谱系在不同地域上的传播表现十分不同。例如,在欧洲(n=71,120),大多数病毒属于L谱系(70,434,99.0%),其中L2d(26,206,36.8%)和L2b(19,416,27.3%)是两个最大的亚谱系,而在亚洲(n=8066),S谱系占到了7.1%(576),L谱系最初以L2d(2381,29.5%)为主,但随后L2g的频率显著增加。其余大洲各个谱系出现的频率也不尽相同。图2结果显示在地理条件、交通运输、文化风俗、防疫政策、超级传播者等各种因素共同作用下,病毒谱系在不同地域之间有各自独特的分布模式,这为病例的分子溯源提供了理论依据。
研究团队还收集了国内从2020年4月到2021年1月的输入病例数据,将它们在谱系划分系统单倍型网络上进行展示(图3),可以为输入型病例的来源提供推断信息。
此外,各个谱系频率的时空变化轨迹还能为理解新冠病毒的演化规律提供线索。第一,虽然取样偏差、奠基者效应可能会造成谱系频率的波动,但那些显著快速在人群中流行的病毒谱系可能是由于其获得了有利突变,据此可以追踪这些突变对新冠病毒临床特性造成的改变,帮助理解其致病机理以及开发潜在的治疗方案。第二,研究者发现病毒基因组上的许多SNV存在极强的连锁性,大大偏离了中性进化的预期,这可能说明这些SNV之间普遍有上位效应存在,亦即病毒出现一个有害突变后,可能会很快产生另一个“代偿性”突变将上一个突变的害处抵消掉。这可能反映了新冠病毒具有“多分枝化”的进化特征,造就了各个谱系之间相当复杂的系统发生关系。
总之,该研究囊括了目前出现的绝大多数新冠基因组变异并整理了它们的谱系关系,阐明其中各个谱系时空分布的规律,搭建出新冠病毒演化的大体框架,对理解病原体变异规律、新冠流行病学追踪、预测病毒的演化方向有重要意义。
北京市疾控中心,中国医学科学院病原生物学研究所,中科院北京基因组研究所、上海营养与健康研究所、分子植物科学卓越创新中心、微生物研究所以及武汉大学等单位参与该课题研究。研究工作获得国家自然科学基金委、科技部以及中科院的支持。
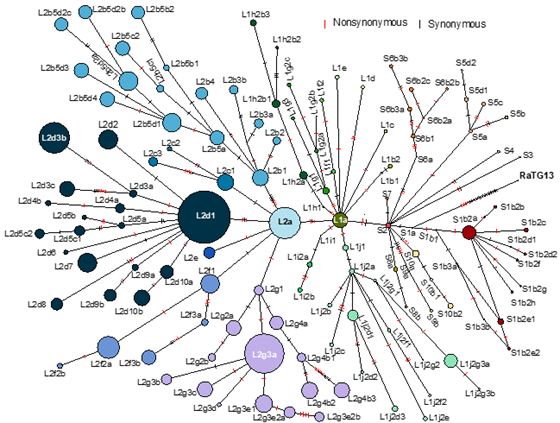
图1. 基于206个分型标记位点的130个亚谱系的单倍型网络
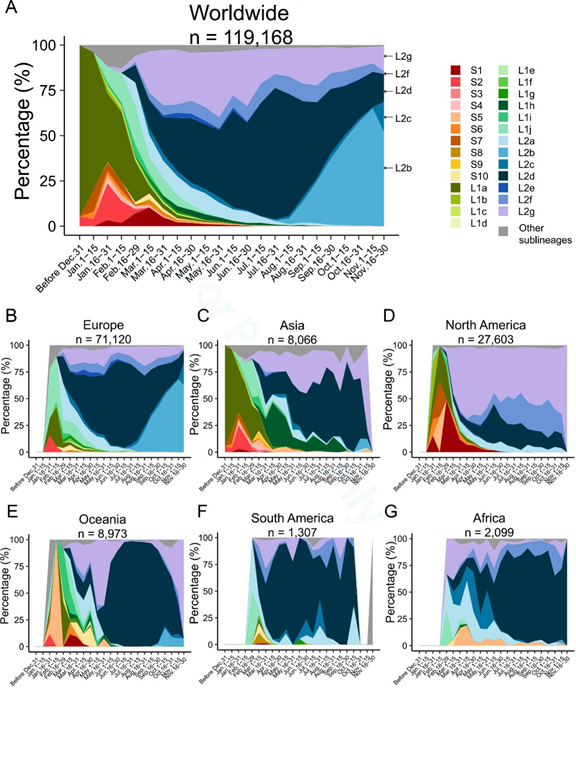
图2. 谱系/亚谱系的时空分布
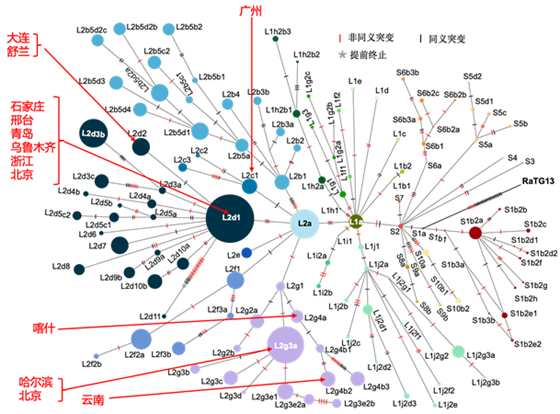
图3. 输入型病例亚谱系的单倍型网络分析


